Research Area
AI for Chemical & Materials Systems
Integrating data-driven modeling, molecular simulation, and artificial intelligence to accelerate the discovery, prediction, and design of chemical and materials systems.
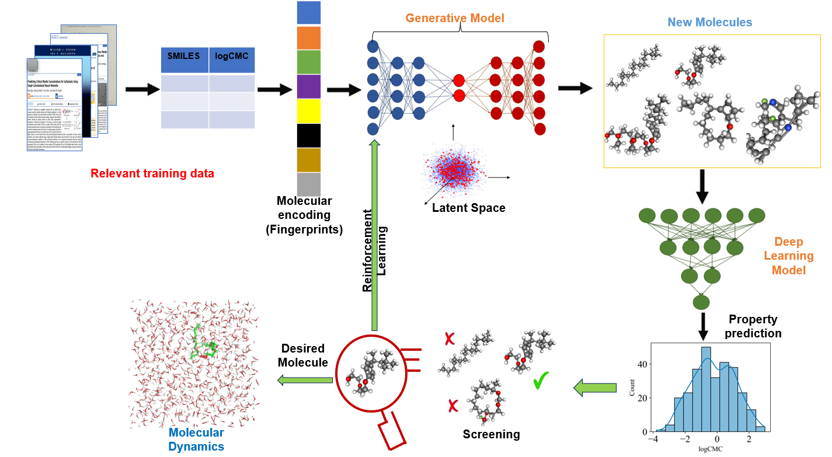
Molecular Design
AI-Driven Materials Discovery
The PSE group develops generative AI workflows for molecular and materials discovery. These methods combine curated experimental datasets, molecular representations, latent-space generative models, predictive property models, and reinforcement learning to propose candidate molecules with targeted physicochemical behavior.
Candidate structures are screened using deep learning and simulation-guided analysis, allowing the design loop to connect molecular generation with property prediction, molecular dynamics, and domain knowledge. This framework supports the identification of promising molecules before experimental testing, reducing trial-and-error in materials development.
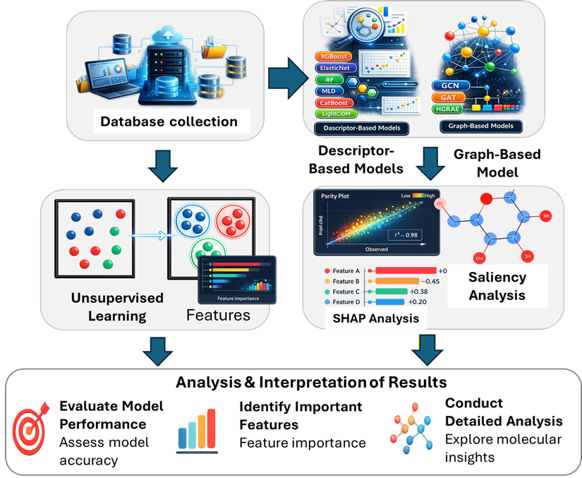
Data & Modeling
Molecular Property Prediction
We build predictive models that connect molecular structure to key material and formulation properties. Our workflows combine database collection, descriptor-based machine learning, graph-based neural networks, and unsupervised learning to identify structure-property trends in complex chemical datasets.
Model interpretation is a central part of this research. Feature importance, SHAP analysis, saliency analysis, clustering, and detailed molecular evaluation are used to understand which molecular features drive model predictions. These insights help transform AI models from black-box predictors into practical tools for chemical engineering decision-making and molecular design.